Michigan State University
Computational Spatial Genomics Lab
Research
Systems-level prediction of long-range gene regulatory networks and the underlying molecular mechanisms
We develop computational and statistical algorithms (probabilistic graphical models, deep learning models etc.) to predict and interpret regulatory networks of gene expression involving sophisticated mechanisms, such as distal enhancers, insulators, and boundary elements, for diverse panels of tissues, cell types and single cells. Multi-omics features (epigenomics, transcriptomics, proteomics) are integrated to delineate the long-range regulatory interactions and to derive systems biology insights.
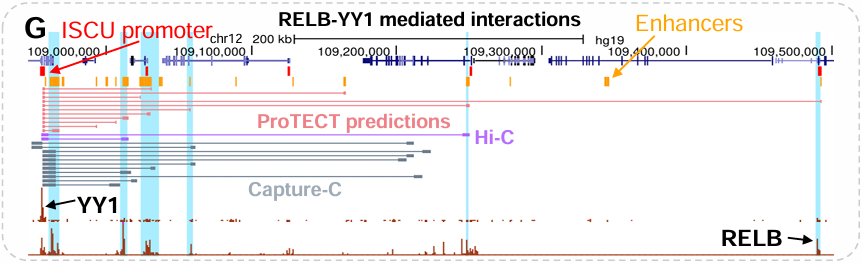
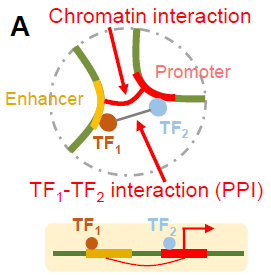
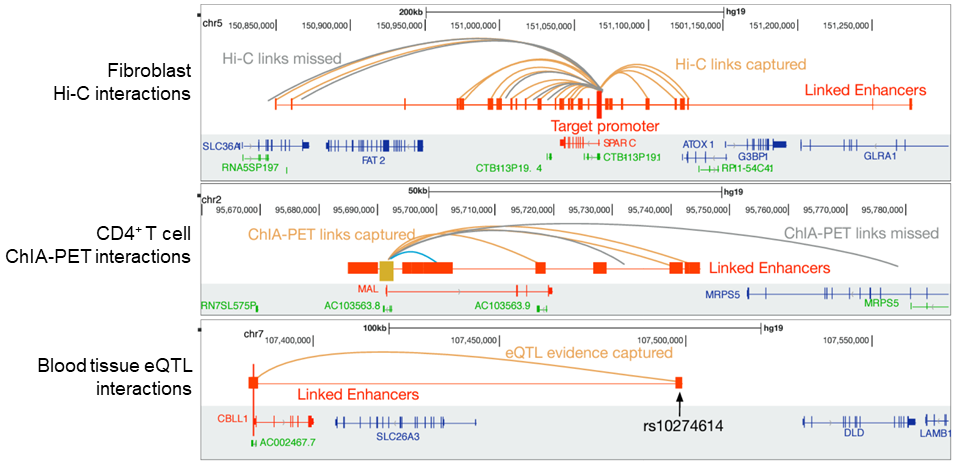
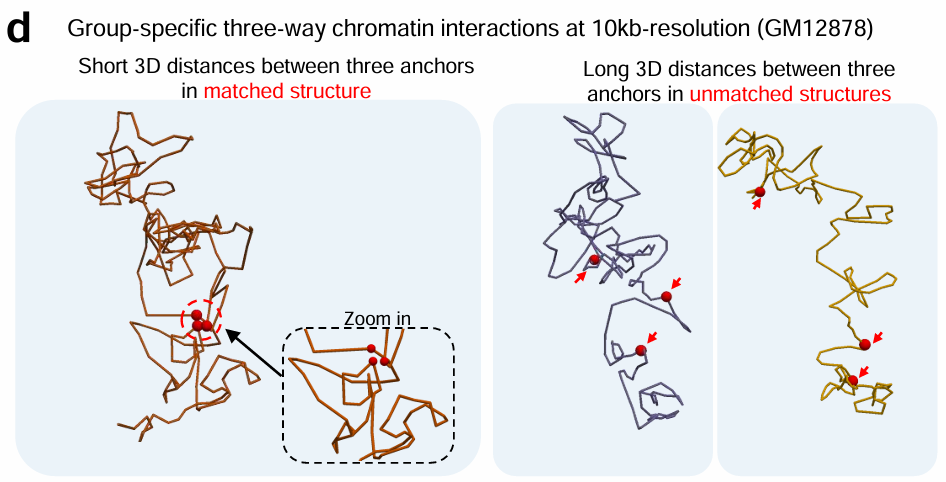
High-resolution reconstruction of spatial genome architectures in 3D space under specific cellular contexts
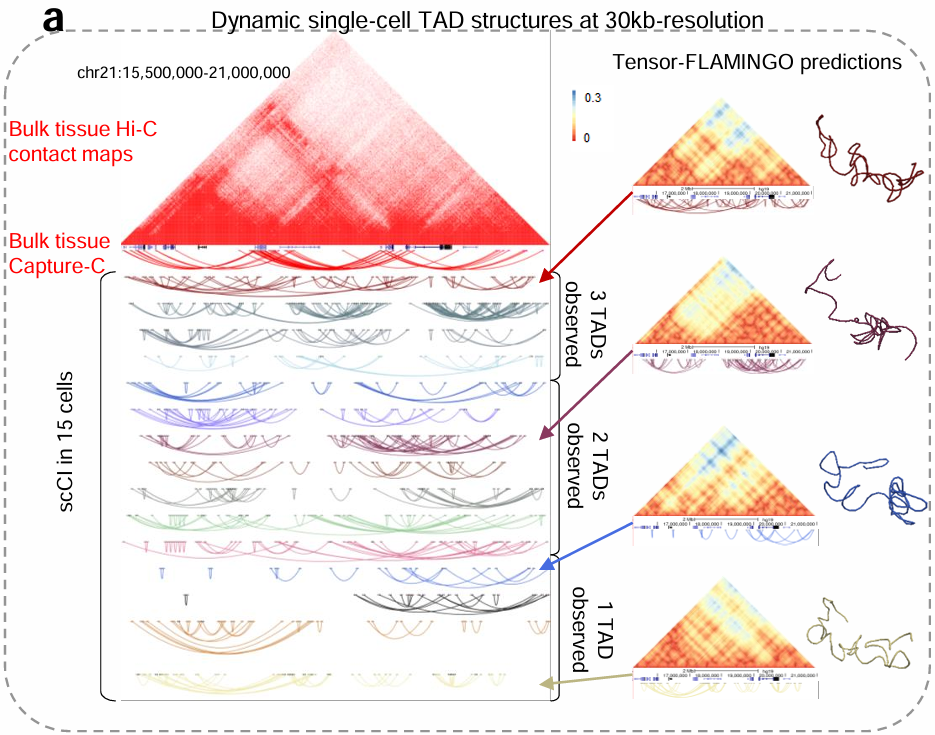
To foster 3D understandings of genome folding, beyond analyses of 1D DNA sequences and 2D chromatin contact maps, we build computational and mathematical models to reconstruct the spatial configurations of genomes at high resolution (e.g. 500bp and 1kb-resolution), based on large-scale Hi-C or related data cohorts. We quantitatively predict the heterogeneous 3D genome configurations across cellular contexts (cell types, sub types, single cells) and analyze the chromatin re-organization patterns at different scales.
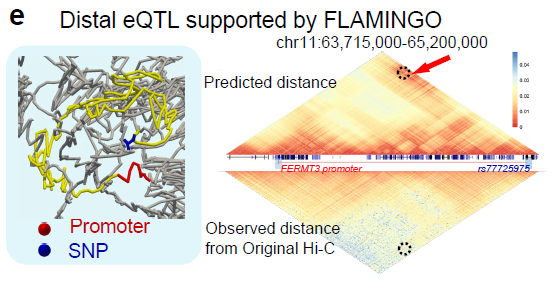
Decipher the functional effects of genetic variants on gene expression and complex traits
We design algorithms to predict the effects of genetic variants, especially distal non-coding SNPs, at the molecular levels (i.e. gene expression) and at the organism levels (i.e. complex traits), by incorporating the regulatory networks.
Building computational infrastructure for high-dimensional biological data
We build computational infrastructure for efficient, scalable and robust analyses of high-dimensional biological data, such as spatial transcriptomics, scATAC-seq, scHi-C, Hi-C, proteomics, ChIP-seq and others.
Software
List of newly developed software packages at MSU that are now released to public. Other newly developed software packages are not included here pending peer-review of manuscripts.
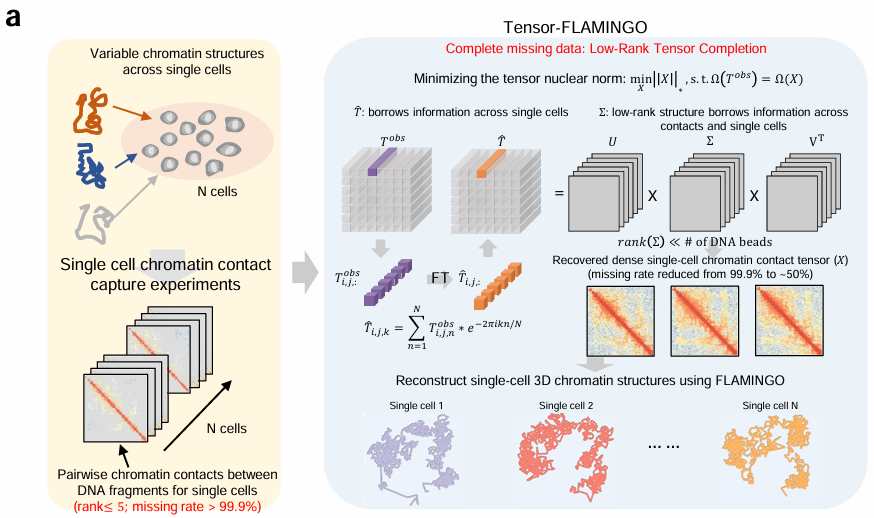
Tensor-FLAMINGO
Single-cell specific high-resolution reconstruction of three-dimensional genome structures based on sparse chromatin contact maps.
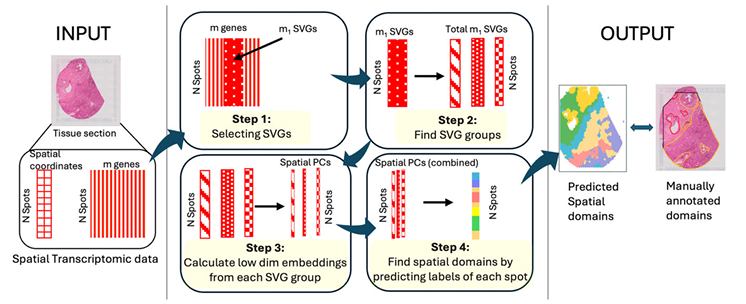
SPACE
Spatial domain detection based on spatially variable gene clustering with cell-type effect adjustments.
FLAMINGO
Three-dimensional genome structure reconstruction algorithm based on chromatin contact maps.
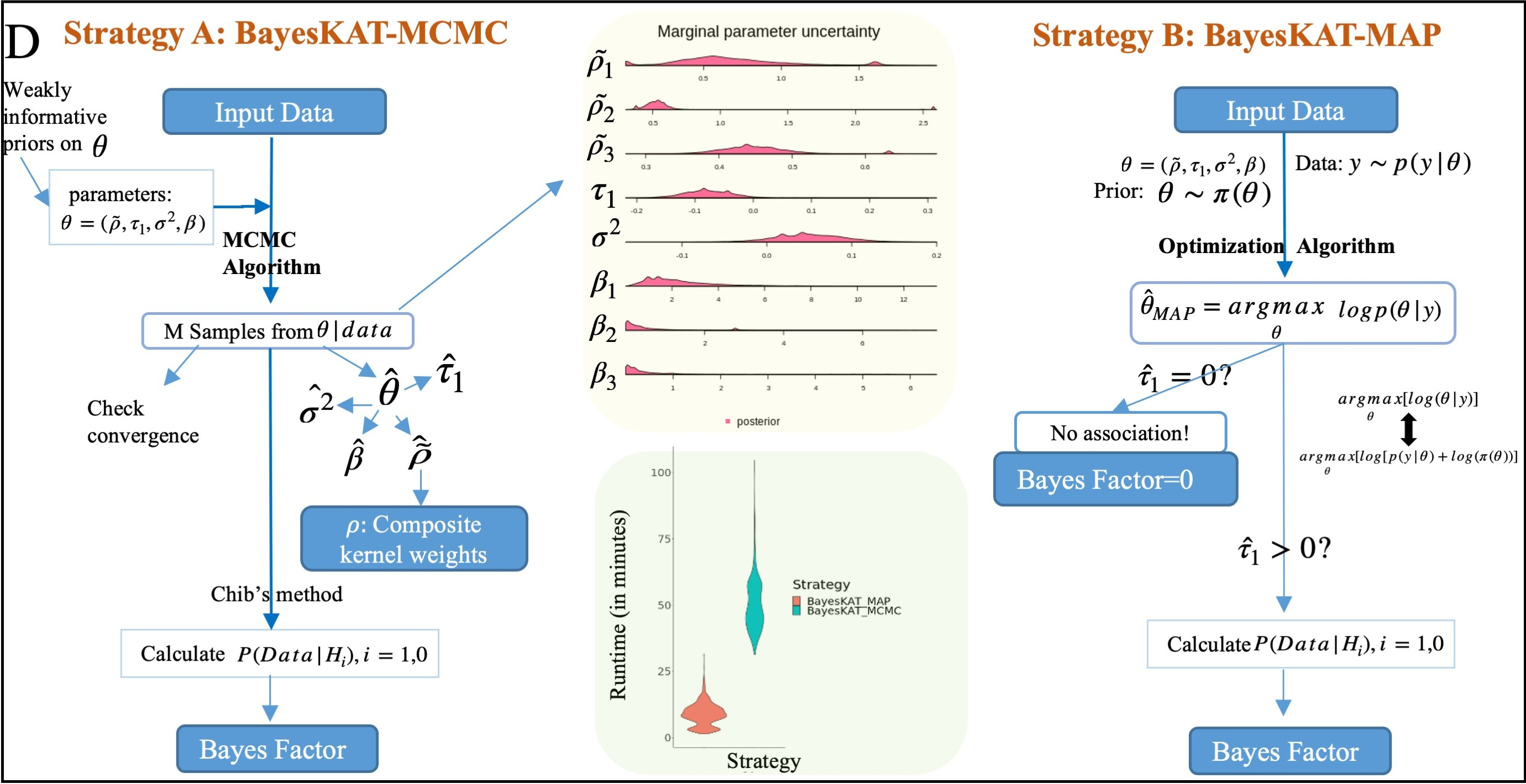
BayesKAT
Bayesian probabilistic model and efficient inference algorithm to automatically select optimal kernel for kernel-based genetic association tests in complex diseases.
ProTECT
Supervised graph-based prediction algorithm for long-range regulatory links based on complex combinatorial PPI signatures.
REGISTER-ME
MCMC algorithm to discover novel functional elements in the human genome for highly repetitive regions.
APRIL
Novel disease-associated gene prediction software based on long-range regulatory networks.
Collaborators
We are excited to work with our collaborators across different scientific domains to understand the biology of functional genomics (immune diversification, genome duplication, repetitive DNA & transposable elements), evolution, molecular genetics, differentiation and development; and to understand the biomedical mechanisms of complex diseases (cancer, Alzheimer's disease, autoimmune diseases etc).
Our current collaborators come from a broad range of institutions:








